

Introduction:Sickle cell disorders were originally found in the African regions, Arabian Peninsula and parts of India. However, in today's age of globalization patients with homozygous or compound heterozygous Sickle cell disorders can be found all over the world. The objective of our study was to assess the distribution and clinical presentation of patients with Sickle homozygous or heterozygous diseases in the Eastern part of India.
Methods:Patients who attended the Thalassemia Clinic in our tertiary care center, between 1st January 2018 to 31st May 2020 (2 years and 4 months) were retrospectively analysed and the ones with a component of Sickle haemoglobin(HbS), either in the form of Sickle cell anemia/homozygous Sickle cell disorders(SCA) or compound heterozygous diseases, like Sickle cell/β thalassemia(HbS/β), Sickle cell/Delta thalassemia(HbS/D), Sickle cell Haemoglobin/E thalassemia(HbS/E), were included in the study. People having Sickle cell trait (HbS trait), have also been included.
Thorough history of painful crises, blood transfusions, family history and treatment history was elicited and every patient was clinically examined. The patients were diagnosed by High Pressure Liquid Chromatography (HPLC) or Thalassemia Mutation analysis by Polymerase Chain reaction (PCR).
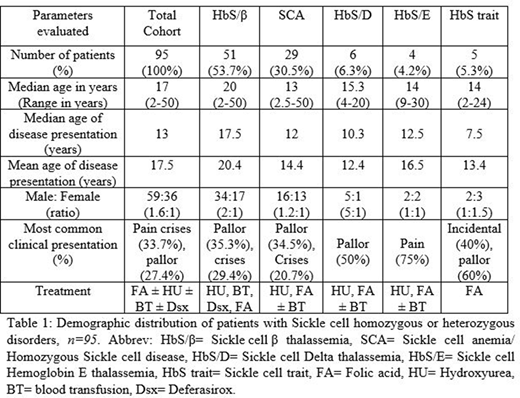
Results:A total of 95 patients with a component of HbS were considered as our study cohort, with HbS/β thalassemia patients being the majority (53.7%), followed by SCA (30.5%). Age of the study cohort ranged between 2-50 years age. HbS/β thalassemia patients presented at a later age (median 17.5 years) than SCA patients (median 12 years). Their demographic distribution is depicted in Table 1.
The most common clinical presentation was painful crisis (32,33.7%), be it abdominal pain (11,11.6%) or bone pain (13,13.7%). Other presenting complaints were pallor (26,27.4%), jaundice (12,12.6%) and fever (4,4.2%). Some rarer presenting manifestations were fatigue (4,4.2%), splenic infarction (1,1%), convulsions (1,1%), Raynaud's phenomenon (1,1%), headache (1,1%) or itchy skin lesions (1,1%). Few patients (4,4.2%) had recurrent pregnancy loss, and one patient was diagnosed incidentally during an antenatal check-up. Most patients had more than one complaint. Very occasionally patients required hospital admission, the reasons being, chest pain, fever, convulsions or abdominal pain.
HbS trait was diagnosed incidentally during evaluation for other illnesses, most commonly during evaluation of pallor (3,60%): one patient was later diagnosed with iron deficiency anemia.
Most patients who attended our center were from within the state or neighbouring states. The patients were treated with Hydroxyurea, with/without blood transfusions, chelation therapy with Deferasirox as required and Folic acid supplementation. People with HbS Trait continued to receive Folate supplementation.
Discussion and Conclusions:This study highlights the varied distribution of HbS among the population attending a tertiary care center, irrespective of a specific area-based population. Till date most studies conducted in India have highlighted the prevalence of Sickle cell disorders among specific focused populations.
HbS/β thalassemia was the most common sickle cell disorder in our study. This is in contrast to most findings in published literature from other countries, where SCA is the commonest. Only one other study conducted in eastern India, has depicted a finding similar to ours.
The median age of disease presentation was at a later age in our study, in contrast to findings in published literature from other countries. There is a variation in the severity of disease manifestation in our study cohort. The most common painful crisis was bone pain, followed by abdominal pain. Pallor was also one of the commonest presenting symptoms. Stroke, a common manifestation of SCA in other countries, was rare in our study cohort.
No relevant conflicts of interest to declare.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal